
BIOTECNOLOGIA
Maximiliano Mendes
A biotecnologia é um campo de estudos
multidisciplinar que combina os conhecimentos obtidos a partir da pesquisa
empírica com os agentes biológicos, como os organismos, células, organelas e
moléculas, a fim de produzir bens e serviços. Por exemplo:
- Obtenção de produtos que promovam a saúde humana ou animal. Ex: produção de insulina e hormônio de crescimento humanos em bactérias geneticamente modificadas.
- Modificar organismos (micro-organismos, plantas ou animais) para que sejam utilizados com fins específicos. Ex: desenvolvimento de micro-organismos capazes de atuar na recuperação de ambientes.
- Desenvolver novos processos industriais. Ex: pesquisas com o intuito de se obter glicose para a fermentação a partir da celulose das células da cana de açúcar, mas em nível industrial, o que pode aumentar a produção de álcool combustível.

Nesse resumo veremos algumas das técnicas e
aplicações utilizadas na biotecnologia contemporânea, porém, é bom destacar que
a biotecnologia nasceu talvez há 8.000 anos, quando o homem aprendeu a produzir
vinhos e pães, mesmo sem ter qualquer noção de microbiologia, biologia
molecular e metabolismo energético. Os procedimentos de biotecnologia vêm então
sendo utilizados pela humanidade desde então, na produção de bens, seleção de
rebanhos e etc. Abaixo se pode ver uma linha do tempo mostrando algumas
descobertas de importância para a biotecnologia.
Na tabela abaixo
estão destacados alguns processos biotecnológicos ou descobertas de interesse
para a biotecnologia e quando foram descobertos. Modificada de Bruno et al. 2014.
Data
|
Processo / Descoberta
|
6000 a.C.
– 2000 a.C.
|
Fermentação
de grãos e cereais.
|
1675
|
Anton Van
Leeuwenhoek observa micro-organismos com um microscópio simples.
|
1876
|
Louis
Pasteur descobre que as leveduras são os micro-organismos responsáveis pela
fermentação.
|
1928
|
Alexander
Fleming descobre a penicilina.
|
1940
|
Fermentação
industrial.
|
1953
|
Descobre-se
a estrutura das moléculas de DNA.
|
1973
|
Início da
engenharia genética.
|
1981
|
Primeira
planta transgênica.
|
1996
|
Clonagem
da ovelha Dolly.
|
2003
|
Projeto
genoma humano completo.
|
Atualmente se executam nos laboratórios uma miríade
de procedimentos que podemos considerar como dentro do âmbito da biotecnologia:
desde preparar uma lâmina para observar algum material no microscópio até
centrifugar uma amostra biológica com vistas a purificar algum componente.
Porém, nosso interesse e foco aqui, serão as tecnologias de manipulação do DNA,
como a tecnologia do DNA recombinante, que objetiva mesclar fragmentos de
moléculas de DNA distintas, podendo ser, inclusive, de diferentes organismos,
gerando DNAs recombinantes. Vejamos a seguir, algumas técnicas, ferramentas e
procedimentos de destaque:
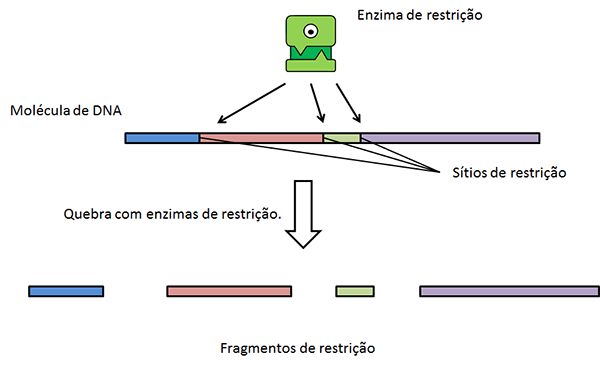
Enzimas de
restrição
Uma das ferramentas mais utilizadas para se gerar
moléculas de DNAs recombinantes são as chamadas enzimas de restrição (ou
endonucleases de restrição), enzimas proteicas encontradas em bactérias e
arqueas, descobertas no final dos anos 1960 e cuja função básica é restringir
as infecções virais: essas enzimas são capazes de quebrar moléculas de DNA em
sequências palindrômicas específicas (ver figura adiante). As bactérias e
arqueas conseguem proteger suas moléculas de DNA da ação dessas enzimas ao adicionarem
radicais metila, -CH3 em citosinas e adeninas na sequência
palindrômica alvo da enzima. A figura a seguir mostra como uma dessas enzimas,
a Eco RI, de Escherichia coli pode
atuar.

Nos pontos de corte das moléculas de DNA, podem ser inserido outros fragmentos, contendo genes de interesse, como veremos posteriormente aqui.
Eletroforese
A eletroforese é uma técnica que permite separar
fragmentos de DNA, como os gerados pelas enzimas de restrição (também é
possível separar proteínas com essa técnica). A ideia básica é a seguinte:
fazer os diversos fragmentos migrarem ao longo de um gel imerso em uma solução,
após posicionar esses fragmentos em uma das extremidades do gel. Esse gel
possui poros, como se fosse uma esponja, por isso permite a migração de
moléculas ao longo dele. Para fazer com que os fragmentos de DNA migrem, basta
submeter esse conjunto a um campo elétrico, de forma que o polo negativo
corresponda à extremidade onde se colocam os fragmentos de DNA, que são
naturalmente negativamente carregados, para que eles migrem em direção a
extremidade oposta, o polo positivo.
Os fragmentos podem ser separados, pois, dependendo
do tamanho, migrarão em velocidades distintas ao longo do gel: os menores
migram mais rapidamente e ao término do procedimento estão mais próximos do
polo positivo, os maiores migram mais lentamente e terminam mais distantes do
polo positivo. A figura a seguir resume o procedimento.

Os retângulos contendo fragmentos de DNA de mesmo
tamanho são chamados bandas. Para observar as bandas é necessário aplicar às
amostras uma substância chamada brometo de etídio (carcinogênica!), que se liga
ao DNA e emite luz se for irradiado com raios UV (também carcinogênicos!).
Essa técnica é usada em diversos procedimentos em
biotecnologia e biologia molecular. Por exemplo, ela permite identificar
pessoas, seja para investigações criminais ou para processos judiciais como os
de confirmação de paternidade. Como? O DNA humano apresenta sequências curtas,
de 9 a 80 pares de bases, chamadas VNTR – Número variável de repetições em sequência (Variable Number of Tandem Repeats).
Essas sequências variam em número de repetições de pessoa para pessoa e também
podem variar entre dois cromossomos homólogos de uma mesma pessoa. Por exemplo,
um indivíduo pode ter em um de seus cromossomos uma VNTR com quatro repetições
e no homólogo, na posição equivalente, a VNTR pode ter seis. Ou então, outro
indivíduo pode ter duas VNTR na mesma posição nos dois homólogos e ainda, um terceiro
ter três VNTR na mesma posição nos dois homólogos. Com essas diferenças em
mente, ao se digerir o DNA de uma amostra qualquer com uma enzima de restrição
que atue delimitando as VNTRs, obtêm-se fragmentos de tamanhos diversos, que
podem ser comparados ao se analisar o padrão de bandas dessas amostras após
utilizar o processo de eletroforese para separar os fragmentos. Como o nome
indica, essas sequências são muito variáveis e, por conta disso, dificilmente
dois indivíduos não aparentados apresentarão as mesmas, então, normalmente o
grau de confiabilidade desses testes é muito alto, com uma precisão acima de 99
%. Veja a figura abaixo.

Clonagem molecular
A clonagem de sequências de DNA consiste em gerar
várias cópias de uma mesma sequência ou fragmento de molécula de DNA, que
inclusive pode conter um gene. Uma forma de se fazer isso é gerar uma molécula
de DNA recombinante e inseri-la em uma bactéria para que seja multiplicada
(clonada) ou algum produto gênico seja produzido.
A técnica básica consiste em utilizar uma enzima de
restrição para cortar um plasmídeo bacteriano e a molécula de DNA que contém o gene
que se quer clonar. Esses plasmídeos são chamados de vetores de clonagem. Os genes
de interesse podem então ser ligados aos plasmídeos vetores e, uma vez
inseridos nas bactérias, esses plasmídeos serão multiplicados (inclusive na
medida em que as bactérias se multiplicam), gerando vários clones com os genes
de interesse. Veja as imagens abaixo:
Esse é o plasmídeo vetor:

O plasmídeo vetor sofre a ação da enzima de
restrição:

A molécula de DNA onde está o fragmento de
interesse também deve sofrer a ação do mesmo tipo de enzima de restrição, de
forma que o corte se dê na mesma sequência alvo e gere extremidades coesivas.

Enfim, o fragmento de DNA de interesse é inserido
no plasmídeo vetor.

Os plasmídeos recombinantes podem ser inseridos nas
bactérias graças ao processo de transformação (lembre-se de que as bactérias
podem receber moléculas de DNA exógenas por transformação, conjugação ou
transdução). O método empregado para se fazer isso é a eletroporação, que
consiste em aplicar um pulso elétrico na solução, criando poros nas células por
onde os plasmídeos podem entrar.
Observando de novo a figura do plasmídeo onde o
gene de interesse será incluído, note que é muito útil, em se tratando do
processo de clonagem com o uso de Escherichia
coli, que os plasmídeos vetores tenham um gene que confira resistência
contra algum antibiótico, como o gene ampR,
que confere resistência contra o antibiótico ampicilina e, assim, permite que
apenas as bactérias que adquiriram o plasmídeo vetor recombinante possam
crescer em um meio de cultura contendo esse antibiótico. A ideia é eliminar as
bactérias que não captaram o plasmídeo vetor, pois elas não geram o produto de
interesse e consomem os recursos do meio de cultura.
Outro gene de interesse, que convém estar presente
em um plasmídeo vetor é o LacZ, que
codifica a enzima beta-galactosidase.
Essa enzima permite às bactérias hidrolisarem o dissacarídeo lactose, ou uma
substância sintética chamada X-gal. Nesse segundo caso, a hidrólise gera um
produto azul de fácil detecção. Novamente, o objetivo é selecionar apenas as
bactérias de interesse, pois a ideia é que o sítio da enzima de restrição
esteja localizado no meio do gene LacZ,
de forma que quando o gene de interesse
é inserido nesse local, o gene da LacZ
é interrompido e deixa de ser funcional. Além disso, as E. coli utilizadas nesse tipo de procedimento têm o gene LacZ defeituoso. Assim, as colônias de
bactérias que receberam o plasmídeo vetor recombinante contendo o gene de
interesse são as que não apresentarem cor azul.
Ainda, é desejável que antes do sítio da enzima de
restrição, onde o gene será inserido, o plasmídeo vetor tenha um promotor muito
ativo, de forma a promover a expressão do gene e a obtenção de seu produto
(lembre-se de que o promotor é a sequência onde o complexo enzimático RNA
polimerase se liga para iniciar a transcrição gênica). Também, é preferível, no
caso da clonagem em bactérias, que o gene clonado tenha sido obtido de um cDNA
(veremos mais sobre eles adiante), pois caso o gene contenha íntrons, não serão
processados adequadamente pelas células bacterianas, visto que elas não possuem
a maquinaria necessária para retirá-los.
Tendo em vista essas limitações, em biotecnologia,
além das bactérias, também se podem utilizar leveduras, que apresentam algumas
vantagens: também podem ser cultivadas com facilidade, possuem plasmídeos, são
capazes de processar pré mRNAs contendo íntrons e efetuar processamento pós
traducional. Para a clonagem em leveduras podem se utilizar plasmídeos ou
cromossomos artificiais de leveduras (chamados YACs – Yeast Artificial Chromosomes). Os DNAs recombinantes também podem
ser inseridos nas leveduras por eletroporação.
Bibliotecas
de DNA e hibridização de ácidos nucleicos
Com o plasmídeo vetor inserido em bactérias, como
fazer para selecionar os clones que contêm o gene de interesse em meio a tantos
outros? O primeiro passo é construir uma biblioteca de DNA, que contém diversos
plasmídeos recombinantes distintos com diversos fragmentos de restrição. Veja a
sequência de imagens a seguir:
Primeiro são gerados os fragmentos de restrição:
Os fragmentos são inseridos em plasmídeos vetores:

Por fim, esses plasmídeos são inseridos em
bactérias, que multiplicam a si e aos plasmídeos: é a biblioteca.

Outro tipo de biblioteca é a de DNA complementar
(cDNA). Nesse caso, extrai-se mRNA das células e com o uso de transcriptases
reversas se obtêm cDNAs. Note que esse tipo de biblioteca representa apenas parte
do genoma, pois não contém os íntrons. Esses cDNAs devem ser modificados com a
adição de sequências contendo sítios de restrição em ambas as extremidades, de
modo que possam então ser inseridos em vetores diversos, como os plasmídeos que
estamos exemplificando. Porém, existem outros tipos de vetores, como o DNA dos
vírus bacteriófagos e os cromossomos bacterianos artificiais (que são plasmídeos
enormes). A figura abaixo resume o procedimento, compare os comprimentos dos
cDNAs (sem íntrons) com os dos genes originais:

Enfim, para detectar o gene de interesse no DNA de
um clone bacteriano é possível usar a técnica de Southern Blot. O procedimento básico consiste em separar fragmentos
de restrição por eletroforese e detectar aqueles de interesse ao permitir que
sequências de DNA cadeia simples chamadas sondas, cuja sequência de bases é complementar
ao nosso gene de interesse venham a se ligar a eles por conta da
complementariedade das bases nitrogenadas. Os passos do procedimento básico são
os seguintes:
- Obtêm-se fragmentos de restrição a partir das moléculas de DNA que se quer investigar (descobrir onde está o gene de interesse).
- Separam-se os fragmentos de restrição de diferentes tamanhos por eletroforese em gel.
- As cadeias de DNA são separadas/desnaturadas ao se submeter o gel à uma solução alcalina de NaOH.
- Comprime-se uma folha de nitrocelulose (negativamente carregada) no gel de forma que alguns fragmentos de restrição desnaturados irão se ligar nessa folha. Outros tratamentos, como o aquecimento, são necessários para que o DNA se fixe adequadamente.
- Incuba-se a membrana de nitrocelulose com os fragmentos de restrição de cadeia simples, aderidos, em uma solução contendo as sondas de cadeia simples para que elas possam se hibridizar, ou seja, ligarem-se de acordo com a complementariedade das bases nitrogenadas. Note que é necessário que saibamos pelo menos parte da sequência de DNA do gene ou do seu mRNA, para que possa haver essa ligação. As sondas podem ser marcadas com isótopos radioativos, substâncias fluorescentes, ou enzimas capazes de gerar produtos fluorescentes para que se possam detectar os clones com maior facilidade.
- Por fim, para saber quais são os fragmentos de restrição contendo o gene procurado (e assim, quais bactérias possuem os clones de interesse) observa-se em quais bandas de fragmentos de restrição separados houve a ligação das sondas marcadas. Isso pode ser feito, por exemplo, caso se tenha usado sondas marcadas com materiais fluorescentes ou radioativos, expondo-se a membrana de nitrocelulose aos raios-X.
A figura abaixo resume o processo.

Organismos
transgênicos
Um organismo que tem algum gene ou fragmento de DNA
de outro organismo distinto, inserido em seu genoma é chamado transgênico. Se
é possível inserir um gene de um organismo distinto, como um humano, em uma
bactéria, por que não ir adiante e tentar inserir genes nos genomas de outros
tipos de organismos, mais complexos, como plantas e animais?
É possível obter mamíferos transgênicos ao se
inserir o fragmento de DNA de interesse no núcleo de uma célula ovo que esteja
sendo fertilizada in vitro. A
primeira etapa desse método é iniciar a fertilização in vitro de uma célula ovo e, durante o processo, inserir os
fragmentos de DNA de interesse no pronúcleo masculino. Alguns dos fragmentos de
DNA de interesse serão inseridos no DNA cromossomal desse pronúcleo, gerando
moléculas de DNA recombinante. Posteriormente, implantam-se os embriões no
útero de uma fêmea e, assim, obtêm-se os animais transgênicos, capazes de
produzir algum produto gênico de interesse.

Para se originar uma planta transgênica, uma
possibilidade é inserir os fragmentos de DNA de interesse em um plasmídeo
chamado Ti (indutor de tumor) da bactéria Agrobacterium
tumefaciens. Essas bactérias são capazes de inserir o plasmídeo Ti no
interior das células das plantas e ele possui uma região capaz de se
inserir no DNA cromossomal, transformar a célula e gerar um tumor. Dessa forma
são geradas várias células com o gene de interesse que podem ser isoladas e
cultivadas em cultura, desenvolvendo-se em plantas transgênicas.


Uma grande facilidade em se trabalhar com os
vegetais, nesse âmbito, é que é possível gerar uma planta completa a partir de
uma ou poucas células vegetais transgênicas. Já com as células animais, esse
procedimento não é possível.
Os procedimentos de geração de organismos transgênicos geram diversas discussões controversas, pois, se por um lado um OGM pode gerar mais de um produto de interesse comercial e até, em tese, diminuir os custos, mas por outro, como estimar quais são os possíveis impactos ambientais que os OGM podem ter? Acredita-se que em alguns casos, os OGM possam ter o metabolismo alterado no sentido de produzir mais de alguma substância tóxica ou que nos cause alergias. Quais populações terão acesso aos produtos? A técnica vai promover o bem estar geral ou acentuar as desigualdades socioeconômicas? São questões ainda não resolvidas.
A reação em
cadeia da polimerase - PCR
A PCR é uma técnica criada em 1985, que permite multiplicar
fragmentos de DNA rapidamente. Tanto a PCR quanto a clonagem permitem multiplicar
fragmentos de DNA, porém, a PCR é mais rápida que a clonagem e mais adequada
para as situações nas quais uma amostra é impura ou contém poucas quantidades
do DNA de interesse a ser amplificado, como por exemplo, DNA retirado de cenas
de crime ou DNA de organismos mortos há muito tempo. Com o uso da PCR se pode
então identificar amostras de DNA de suspeitos de crimes, confirmar se alguém está
infectado por algum vírus, identificar se determinada amostra de DNA pertence a
uma determinada espécie de organismo, amplificar um gene de interesse e etc.
De maneira resumida, a técnica consiste na
repetição das seguintes etapas:
- A mistura contendo os fragmentos de DNA que se deseja amplificar deve ser aquecida até 94° - 96° C, para desnaturar as cadeias de DNA. Nesse caso, a desnaturação consiste na quebra das ligações de hidrogênio que unem as duas cadeias, separando-as. Essa mistura contém também alguns fragmentos curtos de DNA de cadeia simples chamados de iniciadores (ou primers). Os iniciadores possuem uma sequência de nucleotídeos complementar às extremidades do DNA que se quer amplificar (que deve ser conhecido, pelo menos em parte).
- Em seguida a mistura é resfriada até uma temperatura que permita haver a ligação ou anelamento dos iniciadores de cadeia simples, graças à complementariedade de bases, nas extremidades do DNA que se quer amplificar. Essa temperatura pode ser de aproximadamente 50° - 60° C.
- Por fim, a temperatura é novamente elevada e uma DNA polimerase extraída da bactéria Thermus aquaticus cuja temperatura ótima de atividade é por volta de 70° C utiliza as extremidades 3’-OH providas pelos iniciadores ligados para sintetizar novas cadeias de DNA. Ao término, cada molécula de DNA foi duplicada.


Dessa forma, cada ciclo tem o potencial de dobrar a
quantidade de DNA de interesse presente em uma amostra, podendo se obter
milhões de fragmentos em poucas horas.
O sequenciamento
de genomas
O sequenciamento consiste em decifrar a sequencia
completa de todos os pares de bases (pb) de um determinado genoma. O primeiro
genoma completamente sequenciado foi o da bactéria Haemophilus influenzae em 1995 e hoje em dia há vários outros genomas
sequenciados, inclusive os três bilhões de pb do nosso genoma graças ao projeto
genoma humano, completo em 2003. A técnica de sequenciamento básica existe
desde 1980 e é chamada de método de terminação da cadeia com didesorribonucleotídeos
(ou método de Sanger). Esse método consiste em se gerar fragmentos de restrição
e elucidar a sequência de nucleotídeos deles de acordo com os seguintes passos:
- Desnatura-se o fragmento em cadeias simples e eles são adicionados em uma solução contendo oligonucleotídeos iniciadores, DNAs polimerases, os desoxirribonucleotídeos componentes das cadeias de DNA (dATP, dCTP, dTTP e dGTP) e nucleotídeos sintéticos chamados didesoxirribonucleotídeos marcados com substâncias fluorescentes para que sejam detectados facilmente. As DNAs polimerases na solução iniciam a síntese de cadeias de DNA com os monômeros disponíveis.
- A ideia é que cada base nitrogenada presente em um didesoxirribonucleotídeo emita fluorescência de cor diferente. Esses didesoxirribonucleotídeos não possuem nenhum grupo hidroxila (-OH) na pentose, de forma que, sem uma hidroxila, a DNA polimerase não pode continuar a síntese da cadeia de nucleotídeos. Assim, os didesoxirribonucleotídeos terminam a cadeia e geram fragmentos de tamanhos diversos, que emitem luz fluorescente.
- Separam-se os fragmentos utilizando o processo de eletroforese, mas em gel de poliacrilamida (ao invés da agarose, comumente usada para o DNA), pois esse gel forma poros menores e permite separar fragmentos menores e com maior precisão.
- A sequência do fragmento de DNA é decifrada ao se observar a sequencia dos pedaços formados de baixo para cima, ou em outras palavras, os fragmentos menores passam primeiro e os maiores por último. Cada fragmento detectado emite luz fluorescente de uma cor diferente, correspondente à sua base nitrogenada.

Clonagem de
animais
Talvez a técnica mais conhecida para se gerar
clones de animais adultos, como foi o caso da ovelha Dolly em 1997, é o
transplante nuclear. Nesse caso, inicialmente obtêm-se uma célula adulta do
indivíduo que se quer clonar e um ovócito de outra ovelha. Em seguida, retira-se o núcleo do ovócito e
se aplicam pulsos elétricos para fundir as duas células. Dessa forma, obtém-se
um ovócito contendo o núcleo (e outros componentes celulares) da célula adulta
originária do indivíduo que se quer clonar. Depois, aplicam-se mais pulsos
elétricos para promover as divisões desse ovócito. Caso gere um embrião, ele
será implantado no útero de uma terceira ovelha (“mãe de aluguel”), podendo continuar o seu desenvolvimento e chegar à idade adulta.

É interessante notar que os animais clonados a
partir do uso desse método podem vir a apresentar diversos problemas de saúde,
pois além da célula ovo receber componentes citoplasmáticos de uma célula
adulta, já diferenciada, também recebe material genético que já sofreu mudanças
epigenéticas e tem genes inativados.
Assim como nos casos dos organismos geneticamente modificados, a clonagem também gera discussões, pois, se por um lado os clones de algum animal poderiam ter grandes vantagens e se multiplicarem, produzindo mais, por outro, a população de clones perde em variabilidade genética e se torna mais suscetível às adversidades ambientais. Ainda há a discussão sobre a clonagem de humanos, para quê e para quem? De novo, são questões não resolvidas.
Adendos:
A edição gênica com CRISPR-Cas9
Uma técnica recente que tem sido amplamente utilizada na expressão de genes
heterólogos em diversos tipos de organismos, inclusive animais, e tem
facilitado muito esse tipo de trabalho é a que utiliza o sistema bacteriano
chamado CRISPR-Cas9 (de repetições palindrômicas curtas, agrupadas e
interespaçadas regularmente, em associação com a endonuclease 9).
O termo CRISPR se refere a uma região no DNA cromossomal dos procariontes
onde se localizam sequências de DNA de procedência viral (as sequências
espaçadas regularmente). É um registro dos vírus que já infectaram as linhagens
dessas células, mas cujas moléculas de DNA foram cortadas e alguns fragmentos
foram inseridos nessa região. Essas informações são usadas em uma espécie de
sistema de defesa imunitária das bactérias que emprega endonucleases de
restrição, enzimas capazes de cortar moléculas de DNA em sequências palindrômicas
específicas.
As Cas (CRISPR Associated) são endonucleases de restrição cujos genes se
localizam em um operon nas proximidades de CRISPR. As Cas9, por exemplo, formam
um complexo com uma molécula de RNA guia ou rastreadora, sendo que essa
molécula de RNA provém de alguma sequência CRISPR e é usada no reconhecimento e
corte de moléculas de DNA virais. Ao ser infectada por um vírus, a célula
procarionte produz os complexos CRISPR/Cas9 com o RNA guia de sequência
complementar a uma região do material genético viral. Após o reconhecimento e
interação entre as cadeias de RNA associada a Cas9 e o material genético viral,
Cas9 corta o ácido nucleico viral, impedindo a infecção.
No âmbito da biotecnologia, a ideia é usar uma molécula de RNA guia
(sintetizada artificialmente) imitando os transcritos CRISPR e que, associada à
endonuclease Cas9, pode se ligar na molécula de DNA alvo no ponto específico
que se quer cortar. A partir do corte, pode se inserir novas sequências de DNA
na molécula alvo, como um gene, ou então os mecanismos de reparo das células
podem inserir ou deletar (retirar) sequências de nucleotídeos de forma a
inativar um gene. Já foram efetuadas até modificações em embriões humanos com o
uso dessa técnica.

A endonuclease Cas 9 (azul claro) com um RNA guia, que pode ser procedente
das sequências CRISPR ou então, manipulado, de maneira a reconhecer, por conta
da complementariedade de bases, uma sequência em uma molécula de DNA alvo. Uma
vez havendo o reconhecimento, a molécula alvo é cortada e no ponto de corte
podem ser inseridos genes de interesse. Imagem: http://cen.acs.org/articles/92/i39/Patent-Picks-CRISPRCas9-Gene-Editing.html
Referências
Amabis & Martho. Fundamentos da Biologia Moderna. 4ª ed. Moderna. 2006.
Bruno, NA. et
al. Biotecnologia I: Princípios e
Métodos. Artmed. 2014.
Campbell,
Reece et al. Biologia. 8ª ed. Artmed. 2010.
http://biotechlearn.org.nz/focus_stories/transgenic_cows/making_a_transgenic_animal
https://www.labmate-online.com/news/news-and-views/5/breaking-news/the-pros-and-cons-of-genetically-modified-organisms-gmos/31400
http://www.genetica.esalq.usp.br/pub/seminar/BMLima-200901-Resumo.pdfhttp://learn.genetics.utah.edu/content/labs/gel/
http://en.wikipedia.org/wiki/Recombinant_DNA
http://en.wikipedia.org/wiki/Restriction_enzyme
https://en.wikipedia.org/wiki/DNA_profiling
http://www.nature.com/scitable/definition/polymerase-chain-reaction-pcr-110
http://en.wikipedia.org/wiki/Polymerase_chain_reaction
http://en.wikipedia.org/wiki/Whole_genome_sequencing
http://www.sumanasinc.com/webcontent/animations/content/paternitytesting.html
http://en.wikipedia.org/wiki/Southern_blot
http://www.bio.davidson.edu/genomics/method/Southernblot.html
http://croptechnology.unl.edu/animation/GeneGun.swf
http://www.scq.ubc.ca/plant-breeding-versus-plant-genetics/
http://learn.genetics.utah.edu/content/cloning/clonezone/
http://revistapesquisa.fapesp.br/2016/02/19/uma-ferramenta-para-editar-o-dna/
https://www.neb.com/tools-and-resources/feature-articles/crispr-cas9-and-targeted-genome-editing-a-new-era-in-molecular-biology